
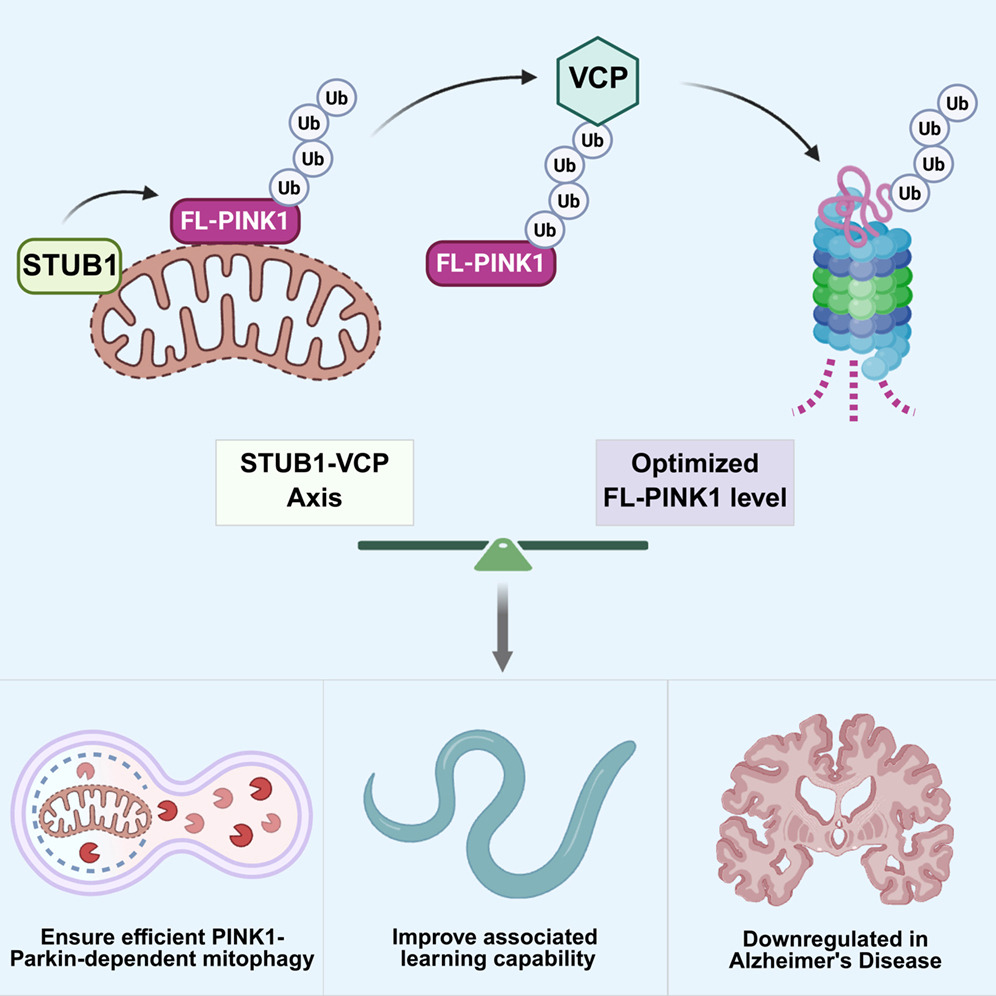
中大新闻网讯(通讯员张玉琦)近日,中山医学院卢广副教授团队联合挪威奥斯陆大学方飞教授团队在期刊《Cell Reports》在线发表题为“STUB1-VCP/p97 complex regulates mitophagy via fine-tuning of PINK1 levels”的研究论文,揭示了一条不依赖于PARL切割的全长PINK1降解新通路,阐明了STUB1-VCP/p97轴通过调控PINK1蛋白水平维持线粒体自噬稳态的分子机制,为理解阿尔茨海默病的发病机理提供了重要理论依据。
研究团队通过经典线粒体损伤模型证实,全长PINK1在损伤后期主要经泛素-蛋白酶体系统降解,且该过程不依赖于传统的PARL蛋白酶切割。利用SILAC定量蛋白质组学技术,团队筛选并验证了泛素E3连接酶STUB1和AAA+ ATP酶VCP/p97是调控全长PINK1稳态的关键分子:STUB1催化全长PINK1发生K48连接型泛素化修饰,随后VCP/p97复合物识别并提取泛素化的PINK1,将其递送至蛋白酶体降解。团队进一步发现,STUB1-VCP/p97轴功能受阻会导致全长PINK1异常积累,这不仅不会增强线粒体自噬,反而产生抑制作用。在分子机制层面,过量的PINK1会过度激活Parkin蛋白,加速其自身泛素化降解,进而导致受损线粒体表面泛素化信号不足及关键自噬受体OPTN和NDP52招募缺陷,最终阻碍损伤线粒体的清除。
该调控轴的生理和病理意义在多层面得到验证。细胞水平上,其缺失会引发线粒体形态和功能异常;在阿尔茨海默病患者海马脑区中,STUB1、VCP/p97及Parkin蛋白水平均显著下降;在秀丽隐杆线虫模型中,该轴的缺失会导致神经元线粒体自噬缺陷和联想学习记忆能力受损。该研究为理解线粒体质量控制机制提供了新视角,也为神经退行性疾病的干预提供了潜在靶点。
中山医学院博士研究生林锦怡、硕士研究生黄泽波、博士后吴勇和挪威奥斯陆大学张诗琦博士为论文共同第一作者,卢广副教授与方飞教授为共同通讯作者。
原文链接:


