
中大新闻网讯(通讯员崔隽)蛋白质是组成生命的物质基础,是生命活动的主要承担者,然而蛋白质在合成之后,往往需要经过一系列蛋白质翻译后修饰(Protein post-translational modifications, PTMs),通过功能基团的添加或去除,调节蛋白质的生化性质从而改变其活性。近日,中山大学生命科学学院崔隽课题组在Molecular Cell杂志83期上连续发表两篇学术论文,分别揭示了蛋白棕榈酰化和焦磷酸化这两种新型PTM对固有免疫应答的关键调控作用机制。
蛋白棕榈酰化介导自噬和NLRP3炎症小体互作调控炎症及其相关疾病的新机制
NLRP3炎症小体是固有免疫系统的重要组成部分,其功能紊乱与自身炎症性疾病、神经性退行性疾病以及癌症的发生发展密切相关。NLRP3突变引起炎症小体的过度激活是早发型炎症性肠病(VEOBD)、幼年特发性关节炎(JIA)和冷吡啉相关周期性综合征(CAPS)等自身炎症疾病的重要危险因素,因此其活化必须被精细调控以维持免疫稳态。崔隽课题组发现S-棕榈酰化通过介导分子伴侣介导的自噬,在调控NLRP3蛋白稳定性中发挥重要功能,并揭示S-棕榈酰化缺陷的NLRP3突变与自身炎症性疾病的发生具有一定的相关性。
S-棕榈酰化是一种新型的蛋白翻译后修饰,参与许多免疫相关蛋白质的转运、定位和稳定性的调节。然而,S-棕榈酰化是否在调控NLRP3炎症小体激活过程中发挥关键作用尚有待阐明。该研究结合S-棕榈酰化抑制剂2-bromopalmitate(2BP)处理、酰基-生物素交换、S-棕榈酰化修饰定点突变、蛋白互作质谱、过表达/敲除棕榈酰转移酶ZDHHC12等实验,证实ZDHHC12可以明显促进NLRP3的棕榈酰化修饰,而其酶活缺失突变ZDHHC12 C127S则不能,表明ZDHHC12促进NLRP3的棕榈酰化修饰依赖于其酶活性。进一步的研究发现Zdhhc12缺陷可在小鼠体内促进NLRP3炎症小体的激活和IL-1β的释放,从而加重Alum诱导的腹膜炎以及LPS诱导的脓毒血症,揭示ZDHHC12在机体中发挥防止NLRP3炎症小体过度激活的重要作用。通过抑制剂实验,研究人员发现溶酶体抑制剂NH4Cl和CQ能够显著抑制ZDHHC12对NLRP3的降解作用。进一步研究发现ZDHHC12介导的S-棕榈酰化修饰通过增强NLRP3与分子伴侣HSC70的结合,促进NLRP3进入溶酶体进行降解,最终抑制NLRP3炎症小体的激活。
为了探究自身炎症性疾病相关的NLRP3突变是否与棕榈酰化失调相关,研究人员筛选了一系列NLRP3突变并发现 NLRP3的D21H、S102L、R490K、和G571R突变可以通过抑制NLRP3的S-棕榈酰化修饰以增强其稳定性,从而促进炎症反应。进一步的机制研究表明,这是由于NLRP3的相应突变降低了NLRP3与ZDHHC12的结合,从而抑制HSC70对NLRP3的识别和降解。由于NLRP3 的S102L突变在亚洲人群中频率较高,且在JIA和CAPS等自身炎症性疾病患者体内被多次发现,该研究将有助于 NLRP3突变相关的自身炎症性疾病患者的诊断与精确治疗。鉴于S-棕榈酰化修饰对NLRP3蛋白激活和稳定性的重要调控作用,特异靶向棕榈酰转移酶的药物可能是治疗自身炎症性疾病的理想靶标。
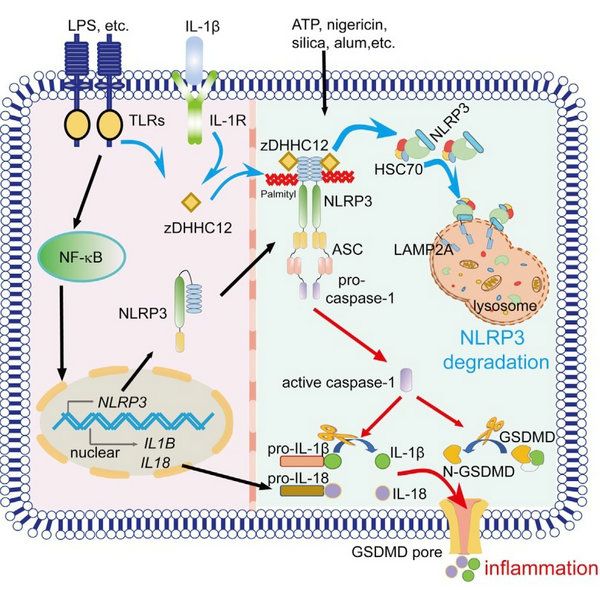
图1 ZDHHC12通过棕榈酰化,诱导NLRP3通过分子伴侣介导的自噬降解,抑制过度炎症反应的工作模型
这篇研究论文的第一作者是中山大学生命科学学院王丽邱博士,她的主要研究方向为炎症小体的免疫调控机制,于2022年入选博士后创新人才支持计划,并在Nature Communications、Cellular & Molecular Immunology等杂志发表多篇一作文章。崔隽教授为该论文的通讯作者。该研究得到国家自然科学基金和博士后创新人才支持计划等经费支持。
蛋白焦磷酸化调控抗病毒固有免疫的新机制
磷酸化修饰是目前已知分布最为广泛的PTM。蛋白质磷酸化的相关研究起步于20世纪初,直到近十年,每年平均仍有1.5万篇相关研究被报道。据估计,真核生物中有将近1/3的蛋白能够被磷酸化。人类蛋白质组中含有10多万个潜在的磷酸化位点。相对于磷酸化修饰,蛋白的焦磷酸化修饰作为一种新发现的PTM,研究历史相对较短。焦磷酸化修饰最早被发现于糖原合成中。由于目前没有蛋白质的焦磷酸化酶的相关报道,一般认为蛋白焦磷酸化属于无酶催化的修饰。崔隽课题组发现焦磷酸化酶UAP1可以结合到转录因子IRF3上,直接正向调控I型干扰素反应,还发现UAP1作为第一个被发现的蛋白焦磷酸化酶,通过催化IRF3的焦磷酸化,增强机体固有免疫应答,降低病毒感染的新机制。
该研究首先通过功能筛选,发现敲除糖基化底物合成中关键的焦磷酸化酶UAP1能够明显抑制细胞的抗病毒活性,而过表达UAP1则会增强I型干扰素以及干扰素刺激基因的表达。Uap1敲低的小鼠对各种病毒的感染都更敏感,其抗病毒能力明显下降,揭示了其在机体抗病毒免疫中的重要作用。为了探究UAP1调控抗病毒免疫的具体机制,研究人员通过免疫共沉淀实验发现UAP1可以特异性结合转录因子IRF3。UAP1的酶活突变体丧失了其促进I型干扰素通路激活的能力,表明UAP1的抗病毒功能依赖于其酶活性。研究人员进一步确定UAP1主要通过催化IRF3的焦磷酸化修饰,而不是糖基化修饰影响IRF3的活性。通过抑制剂实验,研究人员发现O-糖基化抑制剂(OSMI-1)或者N-糖基化抑制剂(TM)处理细胞不会影响UAP1对I型干扰素的促进作用,而焦磷酸化修饰抑制剂(TNP)处理会明显抑制UAP1的抗病毒功能。补回UAP1在己糖胺通路中的重要产物UDP-N-乙酰基葡萄糖胺,不能回复敲除UAP1对IRF3及其抗病毒免疫的调控作用,揭示了UAP1可能直接通过催化蛋白的焦磷酸化发挥功能。进一步的研究通过同位素示踪法、焦磷酸试剂盒检测以及质谱实验等不同实验方法,证明UAP1能够特异性催化IRF3的Ser386位点的焦磷酸化修饰。研究人员构建了Ser386位点失活的S386A突变体,发现S386点突变后IRF3不能进一步活化,IRF3 C端5ST活性位点(Ser396)也不能发生磷酸化,同时IRF3二聚化和入核能力也明显下降,这些结果表明UAP1介导的焦磷酸化修饰对抗病毒固有免疫的调控至关重要。
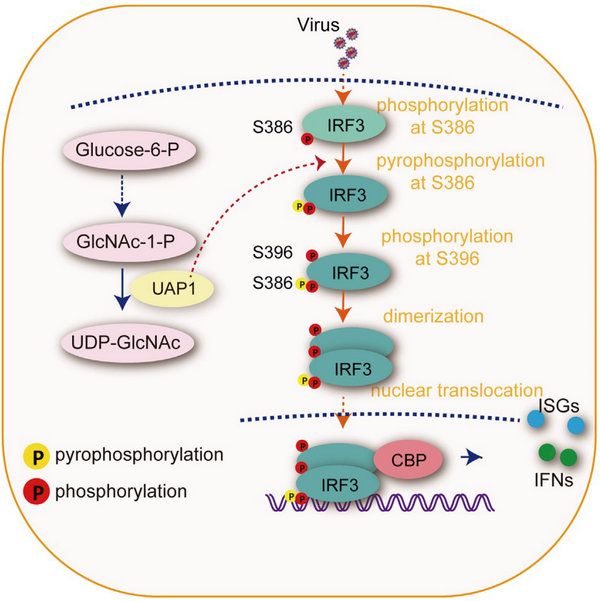
图2 UAP1通过催化IRF3焦磷酸化,调控IRF3活性及其I型干扰素信号通路的工作模型
中山大学生命科学学院崔隽教授为该研究论文的通讯作者,中山大学生命科学学院杨帅博士为第一作者。该研究得到了国家自然科学基金等经费支持。



