
中大新闻网讯(通讯员郑双佳、万园园、杨跃东)蛋白降解靶向嵌合体(PROTACs)作为一种新兴的治疗策略,通过招募泛素-蛋白酶体系统降解致病靶蛋白,具有攻克“不可成药”靶点的巨大潜力。然而,PROTAC分子三元组结构特性导致其较高的分子量进而使其成药性的优化变得极具挑战。国家超算广州中心副总工杨跃东教授团队联合星药科技研发团队,依托“天河二号”凭借其在超算、AI和生物制药的交叉研究及深度融合的经验,充分结合基于强化学习的分子生成和基于物理的分子模拟技术,在短短49天内就发现了具有高降解活性及高药代性能的新型先导化合物,并完成湿实验验证。该研究对PROTAC设计以及成药性优化具有重要意义,相关成果本月正式发表在国际权威期刊Nature Machine Intelligence。
PROTAC聚焦不可成药靶点 其理性设计充满挑战
自2001年首次对蛋白分解靶向嵌合体(PROTACs)进行概念验证以来,PROTACs已成为通过泛素-蛋白酶体系统选择性降解靶蛋白的有效工具。由于其特色的双功能结构,PROTACs有能力同时结合靶蛋白和E3泛素酶,形成一个三元复合物,促进靶蛋白的多泛素化和降解。因此,PROTACs只需要短暂地与目标蛋白结合就能诱导泛素化和降解,这促进了其在“不可成药靶点”上的应用。然而,PROTAC的设计和优化仍需要基于经验主义的反复迭代优化,这种开发策略存在严重的局限性。
PROTAC设计过程示意图。圆圈和线条分别代表配体和连接子,象征着智能模型从广阔的化学空间中提取信息生成一个连接基团将两个蛋白质拉近的过程。
在PROTACs的开发过程中,最棘手的是如何选择合适的连接基团形成合适的PROTAC活性三元复合物从而发挥降解活性和靶点选择性。由于三元结构的复杂性和动态性,连接基团的设计往往是一个艰巨的挑战。连接基团的长度、组成、柔性、连接位点都会对结果造成巨大影响。另外一个设计挑战则来自于PROTAC分子往往不符合口服药物常见的性质。作为一种多组分分子,其较大的分子量导致其相比于传统小分子有着溶解度差、渗透性差、生物利用度低以及不可预测的Hook效应等问题,从而阻碍了PROTACs的临床转化。如何在有限的条件下理性优化PROTAC分子以克服这些缺点,是目前该领域的重大难题。
基于PROTAC-RL优化方法 仅49天发现新型先导化合物
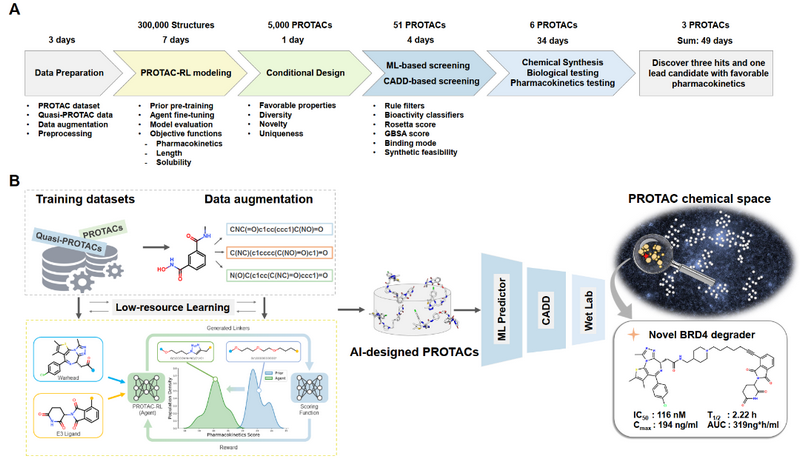
PROTAC-RL流程示意图
为了解决这一问题,研究团队提出了一种基于深度生成模型的PROTAC理性设计算法——PROTAC-RL。该模型以一对E3配体和弹头作为输入,输出设计好的连接物,并在强化学习(Reinforcement Learning, RL)的引导下生成具有特定性质的PROTAC分子。作为概念验证,研究团队选择了BRD4作为靶蛋白并生成了5000余个PROTACs,依托超算通过机器学习打分器和分子动力学模拟方法对这些虚拟分子进行了进一步聚类和筛选。
根据合成可及性,研究者最终挑选、合成并实验测试了6个PROTACs,其中3个显示了对BRD4的抑制性活性。其中一个先导化合物同时显示出对肿瘤细胞系的高抗增殖效力,并在小鼠中表现出良好的药代动力学(如上图所示)。基于国家超算广州中心生物医药应用平台、大规模并行分子动力学模拟方法以及用于深度学习模型训练的海量GPU算力,整个过程仅消耗了49天,证明了超算、深度学习和分子动力学的结合能够促进高效的理性PROTAC设计和优化。
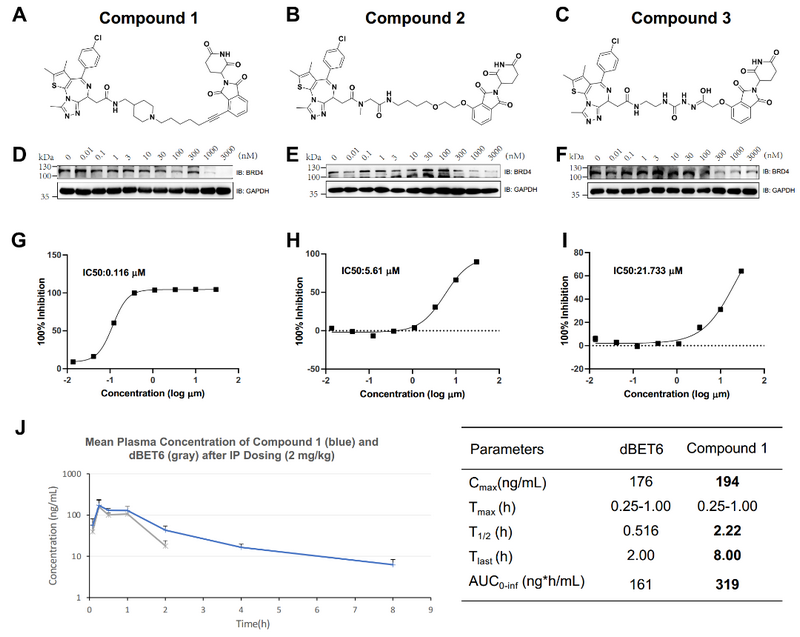
生物活性测试和药代动力学测试
此次研究推出了一种完全自动化的计算框架,该框架有效结合了强化学习驱动的深度生成模型、机器学习和分子动力学模拟,用于合理地设计和优化PROTACs。在针对BRD4靶点的案例研究中,依托“天河二号”及生物医药应用平台,研究团队在不到50天的时间内就发现了三个具有良好降解效应的PROTAC分子,其中一个还具有良好的药代动力学性质,该应用是将超算、人工智能驱动的计算策略与实验相结合获得有效候选药物的典型成果之一。



